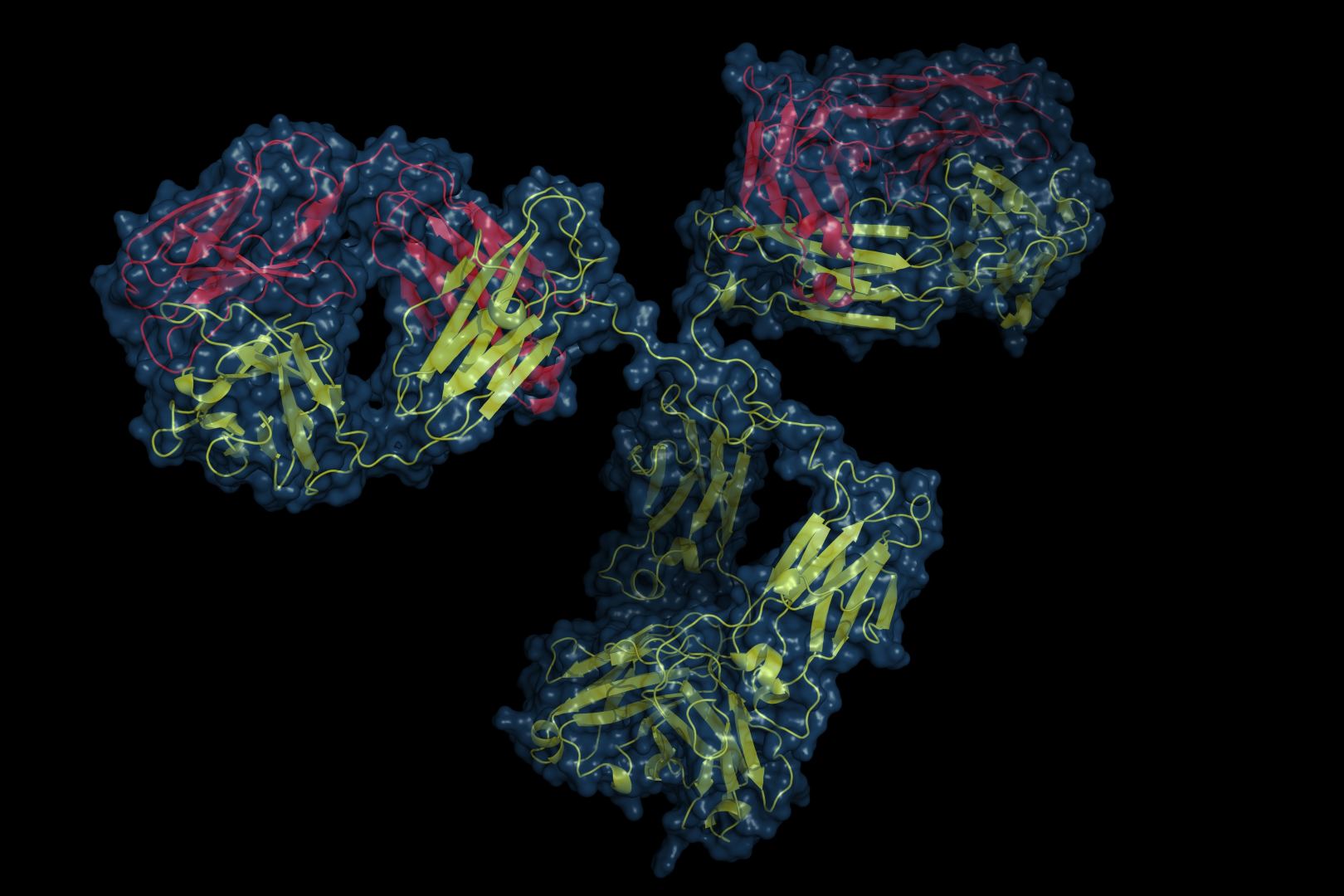
Dr. Č.Venclovas pasakoja, kad baltymai yra viso žmogaus organizmo pagrindas, be jų nebūtų gyvybės. Baltymai sudaro mūsų raumenis, akis, plaukus, jų randama ir skrandyje, o kraujyje esantis hemoglobinas, kuris perneša deguonį, taip pat yra baltymas. Taigi labai svarbu žinoti baltymų struktūras, ypač norint juose ką nors keisti ar slopinti, pavyzdžiui, kuriant naujus vaistus. Eksperimentiškai šias struktūras nustatyti kartais būna labai sunku. Tokiais atvejais gali padėti kompiuterinis modeliavimas. CASP konkurse siekiama patikrinti, ar gali kompiuteriniai modeliavimo metodai konkuruoti su eksperimentais.
„Kai kada gali, o kai kada – ne. Tikslas – galutinai išspręsti šitą problemą. Tai reiškia, kad paėmus baltymo seką kompiuteriniais metodais būtų galima sumodeliuoti tikslią jo struktūrą. Tada struktūros nebereikėtų spręsti eksperimentiškai. Tai padėtų kurti naujus vaistus, taip pat suprasti, kodėl atsiranda viena ar kita liga“, – paaiškina dr. Č.Venclovas.

Kas dvejus metus rengiamas CASP konkursas skirtas pasauliniu mastu įvertinti baltymų struktūrų modeliavimo lygį. Visiems dalyviams duodamos vienodos užduotys – nusakyti tų pačių baltymų struktūrą. Tai daroma aklo testavimo principu. Dalyviai nežino struktūrų, o tai leidžia objektyviai palyginti skirtingų grupių siūlomus baltymų struktūrų nusakymo metodus.
Dr. Č.Venclovas paaiškina konkurso principą: „Mums atsiunčiama baltymo seka ir per dvi ar tris savaites turime pateikti atsakymą, t.y. sugeneruoti baltymo struktūrą, nurodydami baltymo aminorūgščių atomų koordinates trimatėje erdvėje. Visa tai vyksta tris mėnesius. Organizatoriai surenka visų konkurso dalyvių duomenis, kuriuos toliau vertina ekspertai. Jie, remdamiesi savo patirtimi ir kompetencijomis, įvertina ir palygina komandų rezultatus.“
Baltymų struktūros kaupiamos vienintelėje pasaulyje atviroje baltymų struktūrų duomenų bazėje (angl. Protein Data Bank). Joje sukauptos baltymų struktūrų žinios iš viso pasaulio. Mokslininkai čia įkelia sugeneruotas struktūras. Šiuo metu šioje duomenų bazėje galima rasti daugiau kaip 140 tūkstančių baltymų struktūrų, kurios kaupiamos nuo 1976 m. Tačiau, pasak pašnekovo, baltymai, kurių struktūros žinomos, sudaro tik labai mažą dalį visų gamtoje egzistuojančių baltymų. Todėl kompiuteriniai modeliavimo metodai ir yra tokie svarbūs.
VU GMC bioinformatikai CASP konkurse modeliavo baltymų kompleksų struktūras. Tai reiškia, kad jas sudaro dvi ar daugiau baltymų molekulių. Daugeliui procesų būtina, kad baltymai sąveikautų. Tačiau jie taip elgiasi ne atsitiktinai, o sudarydami tam tikrus kompleksus ir tų sąveikų yra gerokai daugiau nei pačių baltymų. Vienas iš VU GMC bioinformatikų tikslų – suprasti šias sąveikas. Tam jie kuria atitinkamus metodus. Pavyzdžiui, jų PPI3D (Protein-Protein Interactions in Three Dimensions) metodas duomenų bazėje padeda surasti struktūrinius šablonus, kuriuos galima panaudoti modeliams konstruoti. Tačiau reikia ne tik surasti šabloną, bet ir įvertinti pagal jį sukonstruoto modelio tikslumą. Tam skirtas kitas VU GMC bioinformatikų sukurtas metodas – VoroMQA (Voronoi tessellation-based model quality assessment).
„Šių metodų kombinacija padėjo tiek sumodeliuoti, tiek atrinkti geriausius modelius ir juos pateikti. Gavę naują seką pasinaudodami PPI3D galime greitai pasižiūrėti, ar yra kas nors panašaus baltymų duomenų bazėje. VoroMQA tinka tiek monomerų, tiek kompleksų struktūroms vertinti. Šiame konkurse pastarasis metodas pripažintas kaip vienas geriausių“, – džiaugiasi VU GMC bioinformatikas.
Mokslininkams baltymo arba jo komplekso struktūros žinojimas leidžia patikrinti hipotezę. VU GMC bioinformatikai, sumodeliavę struktūrą, gali įvertinti aminorūgščių tarpusavio sąveikas. Kitaip tariant, iš baltymų struktūros modelių galima spręsti apie šių baltymų funkciją.
„Modeliai taip pat leidžia iškelti įvairias hipotezes, kurių be struktūrų tiesiog neįmanoma suformuluoti. Kai turi struktūrą, supranti, kaip galėtų atrodyti ir veikti baltymo kompleksas. Tuomet šį mechanizmą galima pradėti tikrinti eksperimentiškai. Vienas iš pastarojo kompiuterinio modeliavimo taikymo pavyzdžių buvo CRISPR-Cas komplekso tyrimas bendradarbiaujant su profesoriaus Virginijaus Šikšnio laboratorija. Kadangi komplekso nepavyko iškristalinti, eksperimentines problemas išspręsti iš dalies padėjo kompiuterinis modeliavimas. Kaip veikia baltymo CRISPR-Cas kompleksas, buvo galima suprasti jau iš kompiuterinio modelio. Pasinaudodami juo, profesoriaus V.Šikšnio laboratorijos mokslininkai atliko eksperimentus, kurie leido daug geriau suvokti, kaip šis kompleksas funkcionuoja, dar prieš sužinodami realią jo struktūrą“, – esminį kompiuterinio modeliavimo privalumą įvardija dr. Č.Venclovas.





